Informations relatives à la réglementation sur les dispositifs médicaux
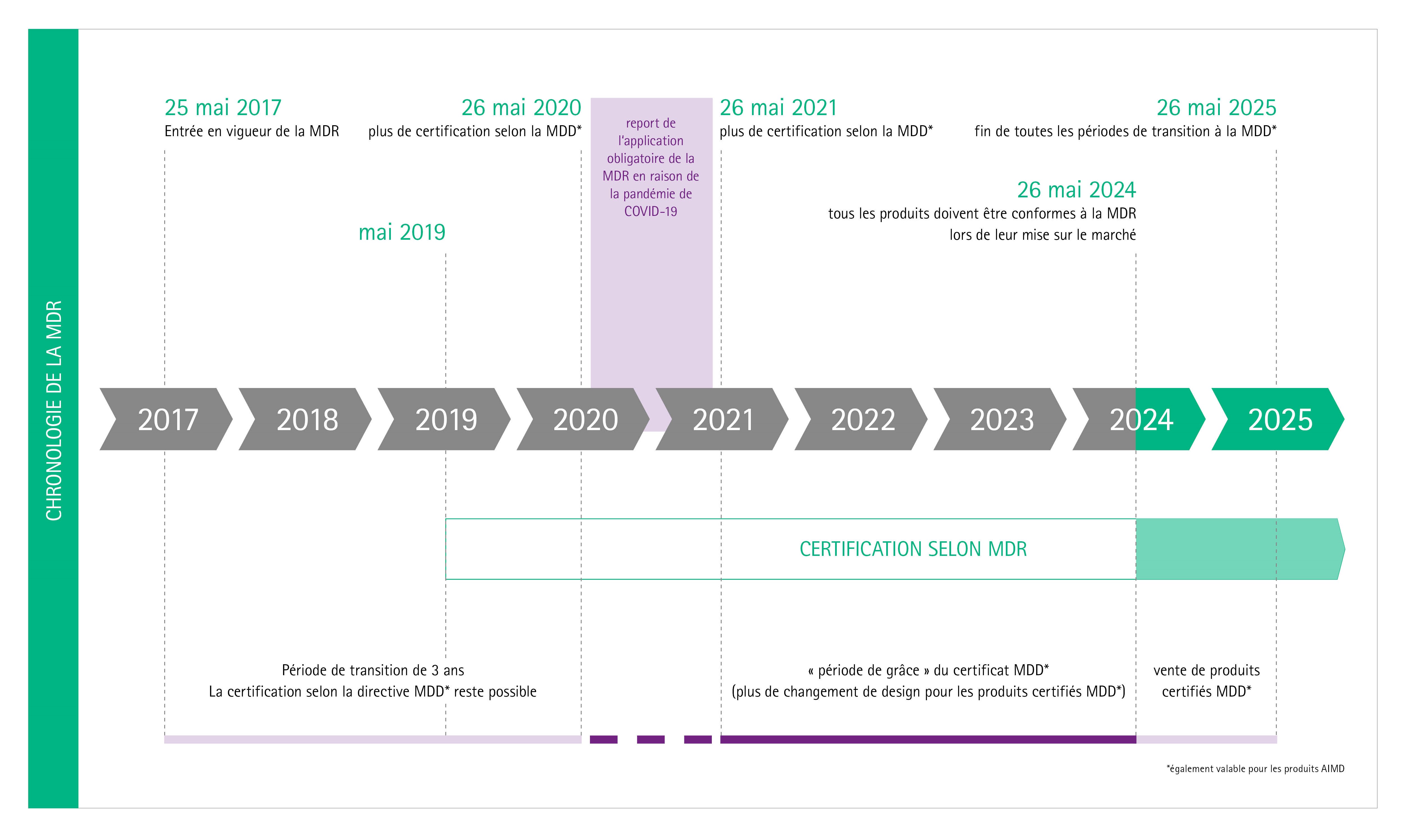
La période de transition pour l’application de la nouvelle réglementation sur les dispositifs médicaux avec ses nombreuses nouvelles réglementations et de nouveaux défis pour toutes les parties concernées, devait prendre fin en mai 2020. La période de transition a été prolongée d'un an en raison de la pandémie de Corona. La nouvelle réglementation est donc entrée en application obligatoire en mai 2021.
B. Braun s'est préparé activement et est en train de mettre en œuvre les nouvelles exigences selon les délais définis par la nouvelle réglementation. Ici, nous avons rassemblé pour vous quelques informations sur la MDR.
La nouvelle réglementation sur les dispositifs médicaux (MDR) est entrée en vigueur en mai 2017. Elle remplace les directives existantes sur les dispositifs médicaux (MDD) et sur les dispositifs médicaux implantables actifs (AIMDD). Toutefois, jusqu'à la fin d'une période de transition qui courrait jusqu’en mai 2021, les dispositifs médicaux pouvaient continuer à être certifiés en vertu des directives précédentes. Pour l'IVDR (règlement sur les diagnostics in vitro), qui remplace l'IVDD (directive sur les diagnostics in vitro) s’applique une période de transition plus longue (5 ans - jusqu'en mai 2022). Sous certaines conditions, cette période peut même être prolongée de 2 ans supplémentaires.
En Europe, on trouve actuellement env. 500 000 dispositifs médicaux qui sont concernés par la MDR. Mais on estime que seuls environ 65 % des dispositifs médicaux seront (re)certifiés en vue de l’obtention du « marquage CE », conformément à la nouvelle réglementation, beaucoup plus exhaustive. Le défi particulier est qu'un nombre décroissant d'organismes notifiés (tel que l'organisme de surveillance technique allemand) devra remplir ces tâches.
Actuellement, la majorité des organismes notifiés sont en phase d'accréditation. On ne sait pas encore ni combien d'organismes de contrôle seront en mesure d'achever la procédure, ni à quel moment ils pourront le faire (liste actuelle de la Commission européenne). En raison des exigences plus élevées à l’égard des organismes notifiés et des fabricants, des ajustements inévitables de portefeuille ont été faits et sont encore à prévoir.
Contexte
Quels sont les domaines réglementaires concernés ?
Voici quelques domaines concernés (liste non exhaustive) :
Règles de classification :
- Les règles ont été étendues pour inclure la classe Ir (instruments chirurgicaux invasifs réutilisables).
- Les exigences relatives aux produits implantables de la classe llb sont plus élevées.
- De nouvelles classes de risque ont été introduites pour plusieurs catégories de produits. Par conséquent, certains produits peuvent être affectées à une classe de risque plus élevée qu’auparavant.
Preuves cliniques :
- À l'avenir, tous les dispositifs médicaux seront soumis aux exigences d'évaluation clinique, quelle que soit leur classe.
Procédures "Scrutiny" :
- Les nouveaux dispositifs implantables de classe III et les dispositifs actifs libérant des médicaments seront contrôlés plus strictement avant leur mise sur le marché.
Organismes notifiés (Notified Bodies) :
- Non seulement, les exigences à l'égard des organismes notifiés sont de plus en plus élevées, mais à l’avenir ils seront également tenus à réaliser des audits inopinés des fabricants.
Documentation technique :
- L’implémentation de la MDR entraîne une importante augmentation du volume de la documentation, et par conséquent, l’effort à fournir par le fabricant croît de manière significative.
EUDAMED :
- Mise en place d’une base de données électronique pour le suivi du cycle de vie des produits.
Quels sont les défis à relever ?
Afin de pouvoir assurer un approvisionnement continu en technologies médicales sûres et innovantes à l’avenir, tous les fabricants sont confrontés au défi de surmonter les exigences accrues en vue de l’obtention du marquage CE.
Les organismes notifiés doivent jeter les bases et créer une capacité suffisante, afin d’être en mesure de réaliser la procédure d'évaluation de la conformité. On peut déjà observer que les périodes désignées de transition pour l'implémentation de la MDR sont étroites et que l'accréditation des organismes notifiés, l'harmonisation des standards existants ainsi que de la structure de la base de données Européenne EUDAMED requièrent davantage de temps. Actuellement, il est probable que cela ne puisse pas être réalisé dans le délai impératif annoncé pour la mise en œuvre de la MDR, en particulier pour la base de données EUDAMED.
Très tôt déjà, B. Braun a lancé des préparations de grande envergure en vue de la certification de ses propres dispositifs médicaux conformément à la MDR. Bien évidemment, cela s'applique à tous les produits B. Braun qu’ils soient fabriqués par B. Braun ou achetés par B. Braun sur le marché dans le but de compléter sa gamme de produits. Au vu de l'avancement des mesures prises, B. Braun est confiant de pouvoir se conformer aux exigences de la MDR.

FAQs
FAQs générales au sujet de la MDR
Pourquoi a-t-il été nécessaire d’implémenter une nouvelle réglementation sur les dispositifs médicaux ?
Une révision au niveau européen s'est avérée nécessaire pour la directive 93/42/CEE relative aux dispositifs médicaux (MDD), qui fut publiée en 1993 et qui est toujours en vigueur aujourd'hui pour les dispositifs médicaux certifiés avant le 26 mai 2021.
Avec cette nouvelle réglementation, les autorités de l'UE souhaitent améliorer la qualité et la sécurité des dispositifs médicaux, harmoniser les processus dans toute l'UE et améliorer la sécurité des patients.
D’autres aspects sont l'amélioration de la transparence et de la traçabilité, en lien avec les nouvelles technologies, ce qui permet une identification précise de chaque produit tout au long de sa durée de vie.
Qu'est-ce qui est régi par la règlementation sur les dispositifs médicaux (MDR) ?
La MDR définit les exigences auxquelles un fabricant doit se conformer pour pouvoir vendre des dispositifs médicaux en Europe. Sont concernées aussi bien les exigences techniques d'un produit que les exigences relatives à la surveillance des produits utilisés dans les établissements de santé.
Contrairement à la directive précédente (MDD), il s'agit d’une réglementation européenne qui s'applique directement dans tous les pays européens.
En raison de la pandémie de COVID-19, la période de transition pour l'application obligatoire de la MDR a été prolongée jusqu'en mai 2021.
Quels sont les principaux changements résultant de la réglementation sur les dispositifs médicaux (MDR) ?
Il y a plusieurs changements dans la classification des dispositifs. Outre l'introduction de la nouvelle classe Ir pour les instruments chirurgicaux réutilisables, ce sont surtout les exigences relatives aux dispositifs implantables de la classe IIb qui sont plus élevées.
De plus, de nombreuses catégories de produits ont été affectées à une classe de risque plus élevée.
La MDR augmente les exigences en matière de données cliniques pour les dispositifs médicaux. À l'avenir, tous les dispositifs médicaux, quelle que soit leur classe de risque, devront faire l'objet d'une évaluation clinique.
La procédure "Scrutiny" nouvellement introduite permet d'améliorer la surveillance avant commercialisation des nouveaux dispositifs implantables de la classe de risque III, ainsi que des dispositifs contenant des médicaments de la classe IIb.
En plus des exigences accrues pour le fabricant, des règles plus strictes pour les organismes notifiés s'appliquent également avec effet immédiat.
Pour obtenir l’autorisation à approuver les dispositifs médicaux, diverses exigences supplémentaires doivent être remplies. En outre, les organismes notifiés sont tenus d'effectuer des audits inopinés dans les entreprises des fabricants.
En raison des exigences supplémentaires concernant la documentation technique à fournir par les fabricants, le volume et la complexité de la documentation augmentent considérablement.
Qu'est-ce que l'EUDAMED ?
Les données pertinentes pour le public seront mises à disposition dans une base de données européenne centrale qui existe déjà aujourd'hui. La version améliorée d'EUDAMED était prévue d'être lancée à la mi-2022. Ce délai est actuellement reporté à la mi-2024.
Dans l’EUDAMED, les fabricants importateurs ou représentants légaux de dispositifs médicaux destinés à être distribués dans l'UE, devront fournir des données sur leur rôle en tant qu’acteur économique, ainsi que des données relatives à chaque produit destiné à être distribué dans l'UE.
La MDR concerne-t-elle tous les produits ?
Oui, tous les dispositifs médicaux des différentes classes de risque, y compris les systèmes et nécessaires (kits) de traitement, sont concernés.
Les dispositifs de diagnostic in vitro, dont la commercialisation est réglementée par le nouveau règlement sur les dispositifs de diagnostic in vitro (IVDR) sont également concernés.
Quel est le calendrier de mise en œuvre de la MDR ?
Suite à la publication de la MDR le 5 mai 2017, le règlement est entré en vigueur le 25 mai 2017 avec une période de transition qui devait durer jusqu'au 26 mai 2020.
En raison de la pandémie de COVID 19, la mise en œuvre obligatoire de la MDR a été reportée à mai 2021.
Jusqu'au 26 mai 2024, les certificats MDD seront toujours valables (par exemple pour les produits des classes de risque II et III).
Toutefois, dans certains cas, il est obligatoire de remplacer le certificat MDD par un certificat MDR (par exemple pour les produits de la classe de risque I).
Dès le 26 mai 2025, les produits dotés d'un certificat MDD ne pourront plus être mis sur le marché.
Quelles classes MDR ont leur date d'entrée en vigueur à partir de quand ?
En raison de la pandémie de COVID-19, l'application obligatoire de la MDR a été effective au 26 mai 2021. Il en résulte les délais suivants pour la mise sur le marché des dispositifs médicaux, par classe de produits :
- Classe I : 26 mai 2021
- Classe Ir, s, m, Classe IIa, Classe IIb et Classe III : 26 mai 2024
Qu’est-ce que l’évaluation de la conformité ?
L'évaluation de la conformité indique si un produit et son fabricant respectif répondent aux exigences de la MDR européenne. En fonction de la classification des risques des produits concernés, B. Braun est autorisé à effectuer elle-même cette évaluation (classe I).
Les évaluations pour les classes plus élevées (classe Ix, IIa, IIb, III) sont effectuées par un "organisme notifié". Comme auparavant, il existe trois classes de risque. Une nouvelle sous- classe de risque "Ir" a été introduite pour les instruments chirurgicaux réutilisables. La déclaration de conformité est la condition nécessaire pour obtenir un marquage CE.
Qu'est-ce qu'un organisme notifié ?
Un organisme notifié est une société privée qui est, après accréditation, "notifié" (c. à d. mandatée) au nom de l'Union Européenne pour vérifier la conformité d'un fabricant avec la MDR. Les organismes notifiés actifs pour B. Braun Melsungen AG sont p.ex. TÜV SÜD, MedCert, Dekra.
Quel marquage obtiennent les produits certifiés MDR ?
Avec l'introduction de la MDR, un nouveau symbole "MD" pour les dispositifs médicaux sera introduit et appliqué sur les étiquettes.
Quels efforts supplémentaires restent à fournir après la fin de la période de transition, le 26 mai 2024 ?
En raison des exigences accrues de la MDR, les fabricants devront faire face à des coûts supplémentaires importants même lorsque l’ensemble de leurs produits sera conforme à la MDR.
Quelle valeur ajoutée la MDR apporte-t-elle aux fabricants, aux prestataires de soins de santé et aux patients ?
Les principaux objectifs de la réglementation sont une meilleure protection de la santé publique et de la sécurité des patients, plus de transparence, une sécurité juridique et un concept plus orienté vers l'Europe. Ces objectifs doivent être atteints grâce à une documentation technique plus complète sur les produits concernés dans un système de gestion de la qualité conforme à la réglementation MDR.
FAQs au sujet de la MDR & B. Braun
Dans quelle mesure B. Braun est-t-elle concernée par la MDR ?
En tant que fabricant de dispositifs médicaux, B. Braun a dû se conformer aux exigences d'ici mai 2021. Différents groupes de travail mettent à jour la documentation technique et révisent les processus pour les rendre conformes à la MDR.
B. Braun est également tenu de fournir à EUDAMED des informations sur les produits, notamment les données d'identification des produits (UDI) ainsi que des informations relatives à la surveillance du marché.
Quels produits B. Braun sont concernés ?
Tous les dispositifs médicaux et les diagnostics in vitro. Également tous les produits qui sont certifiés pour la première fois selon la nouvelle classe Ir.
B. Braun est-il en mesure de respecter le calendrier prévu ?
Cela fait longtemps que B. Braun se prépare activement afin d’être en mesure de se conformer à la nouvelle réglementation selon le calendrier prévu.
Tous les dispositifs médicaux de B. Braun seront-ils certifiés conformément à la MDR ?
En fonction du cycle de vie prévu, les produits seront certifiés selon la MDR. A la fin de la période de transition, c.à.d. au plus tard en mai 2024, l’ensemble des produits B. Braun sera conforme à la MDR.
Pendant la période de transition, B. Braun commercialisera à la fois des produits certifiés MDD et MDR.
Les organismes notifiés mandatés par B. Braun répondent-ils aux exigences futures ?
En mai 2019, TÜV Süd fut le deuxième organe de contrôle accrédité en tant qu’organisme notifié pour le monde entier. D'autres organismes notifiés qui inspectent les dispositifs médicaux de B. Braun ont également été désignés conformément à la MDR ou sont en phase d'accréditation. Une vue d'ensemble des organismes notifiés conformément à la MDR est disponible sur le site web de la Commission européenne.
FAQs au sujet de la MDR & B. Braun Medical Suisse
Informations sur le MRA (Mutual Recognition Agreement)
La réglementation relative aux dispositifs médicaux (MDR) est entrée en application obligatoire le 26 mai 2021. Pour garantir un statu quo dans le processus d’importation en Suisse de dispositifs médicaux en provenance de l’UE, l’accord de reconnaissance mutuelle (MRA) signé entre l’Union européenne (UE) et la Suisse (CH) devait être actualisé avant le 26 mai 2021. Le MRA n'a malheureusement pas été actualisé et par conséquent l'ordonnance sur les dispositifs médicaux a été adaptée pour clarifier les règles pour les importations à partir du 26 mai 2021. De plus la Suisse n'a plus accès à la base de données EUDAMED et est en train de développer une base de données équivalente qui sera appelée SWISSDAMED.
B. Braun Medical SA a donc endossé le rôle de mandataire suisse (CH-REP) pour éviter toute pénurie de dispositifs médicaux B. Braun importés de l'UE. B. Braun Medical SA joue également le rôle de mandataire suisse pour certains dispositifs médicaux achetés à des fabricants tiers.
Par cette mesure, nous pouvons confirmer que tous les produits B. Braun sont toujours disponibles sur le marché suisse depuis l'échec de la MRA avec l'UE le 26 mai 2021.